

Abstract
Chimeric antigen-receptor (CAR) engineered T cells have mediated impressive outcomes in a subset of hematological malignancies, yet this therapy remains highly personalized and largely ineffective against more widespread epithelial and solid tumors. Cellular manufacturing processes tailored to individual patients, and an inability to overcome the complex and immunosuppressive properties of the solid tumor microenvironment remain formidable challenges in the effort to achieve widespread application of adoptive cell therapies for cancer. Genome editing strategies using targeted nuclease platforms are being developed to overcome these limitations, and several are already entering clinical application. Multiplex gene editing strategies to develop off-the-shelf cellular therapies against non-hematological malignancies are of particularly high interest but remain limited by concerns surrounding off-target nuclease activity and chromosomal translocations induced by simultaneous double-strand break (DSB) induction at multiple genomic loci. The risk of genotoxic side-effects is further amplified when combining multiplex DSB induction with randomly integrating platforms for antigen-specific receptor delivery.
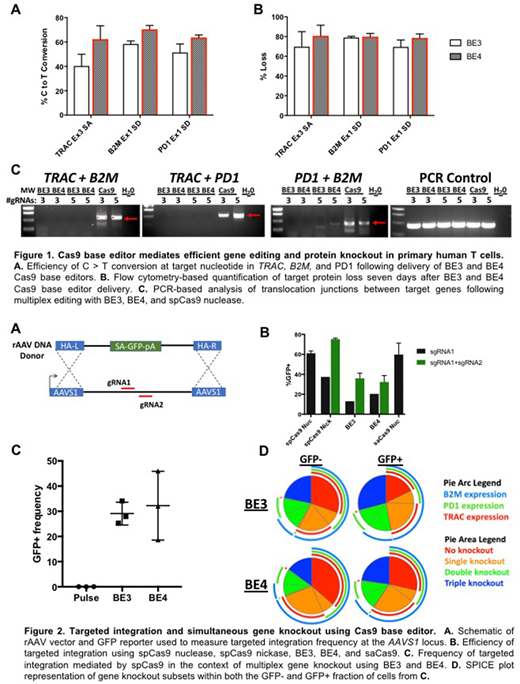
An ideal strategy would allow for multi-gene disruption and targeted integration of antigen-specific receptors without introducing multiple genomic DSB. To this end, we evaluated the application of third- and fourth-generation Cas9 base-editor technologies for gene disruption in primary human T cells. Through systematic reagent and dose optimization efforts we achieved highly efficient, targeted cytosine to thymine (C > T) base conversion (Figure 1A) and consequent protein knockout (Figure 1B) at multiple therapeutically relevant loci including TRAC (KO = 80.8%, SD = 10.7%), PD1 (79%, SD = 4%), and B2M (80%, SD = 3%). We observed that fourth generation base editor (BE4) achieved consistently higher C > T conversion rates with reduced non-canonical editing (i.e. C>A, G) overall compared to third generation (BE3). Targeted disruption of splice acceptor (SA) and splice donor (SD) sites resulted in higher frequency of protein knockout versus induction of premature stop codons at all loci examined.
Importantly, while multiplex editing of these loci using Cas9 nuclease resulted in detectable translocations between all targeted sites, these translocations were largely eliminated using BE4 (Figure 1C). Finally, we exploited both the single strand nickase function of BE4 and orthogonal Cas enzymes in conjunction with rAAV delivery to achieve targeted integration of a gene expression cassette at the AAVS1 safe harbor locus (Figure 2A,B). Targeted integration using an orthogonal Cas9 system (SaCas9) in conjunction with multiplex base editing achieved simultaneous gene knockin and gene knockout (Figure 2C). Engineered T cells expand to clinically relevant numbers and maintain cytokine polyfunctionality and the ability to kill target cells when equipped with a chimeric antigen receptor. Collectively, we demonstrate that Cas9 base-editor technology can be utilized to mediate efficient, multiplex gene disruption and targeted gene integration in primary human T cells without associated translocations. This streamlined approach to genome engineering may be broadly applied for the development of safe and effective cell therapies.
Webber:BEAM Therapeutics: Consultancy; B-MoGen Biotechnologies: Employment, Equity Ownership. Moriarity:B-MoGen Biotechnologies: Employment, Equity Ownership; BEAM Therapeutics: Consultancy.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal